Scaling deep learning for materials discovery
논문 정보
- 제목: Scaling deep learning for materials discovery
- 저자: Amil Merchant (Google DeepMind), Simon Batzner (Google DeepMind), Samuel S. Schoenholz (Google DeepMind), Muratahan Aykol (Google Research), Gowoon Cheon (Google Research), Ekin Dogus Cubuk (Google DeepMind)
- 학회/저널: Nature
- 발행일: 2023-11-29
- DOI: 10.1038/s41586-023-06735-9
- 주요 연구 내용: 본 연구는 그래프 신경망(GNN)을 대규모로 확장하고 액티브 러닝을 적용하여 무기 결정의 안정성을 정확하게 예측하는 GNOME(Graph Networks for Materials Exploration) 프레임워크를 개발했다. 이 프레임워크는 대칭성을 고려한 부분 치환(SAPS)과 같은 새로운 후보군 생성 방법과 밀도 범함수 이론(DFT) 계산을 결합한 반복적인 학습 사이클을 통해 모델을 지속적으로 개선하며 방대한 화학 공간을 효율적으로 탐색함.
- 주요 결과 및 결론: GNOME을 통해 기존 연구 대비 220만 개의 새로운 안정적인 결정 구조를 발견했으며, 이 중 381,000개는 새로운 볼록 껍질(convex hull)을 형성하여 인류에게 알려진 안정적인 재료의 수를 10배 가까이 확장함. 최종 모델은 에너지 예측 오차를 원자당 11 meV까지 낮췄으며, 안정적인 구조 예측의 정확도(hit rate)를 80% 이상으로 향상시킴. 또한, 이 과정에서 생성된 방대한 데이터셋은 전이 학습 없이도 높은 정확도를 보이는 범용 머신러닝 원자간 전위(MLIP) 모델 개발을 가능하게 함.
- 기여점: 알려진 안정적인 무기 재료의 수를 10배 가까이 확장하여 재료 과학 분야에 방대한 데이터를 제공함. 딥러닝 모델의 규모를 확장함으로써 학습 데이터 분포를 벗어나는 문제(out-of-distribution)에 대한 일반화 성능이 향상될 수 있음을 보여주었고, 이는 과학적 발견에서 머신러닝의 근본적인 한계를 극복할 가능성을 제시함. 또한, 생성된 데이터셋을 통해 특정 재료에 대한 추가 학습 없이도 분자 동역학 시뮬레이션에 바로 사용될 수 있는 고성능 사전 학습 원자간 전위 모델을 개발함.
요약
초록
기존의 재료 발견은 비용이 많이 드는 시행착오 접근법에 의해 병목 현상을 겪어왔다. 본 연구는 대규모로 학습된 그래프 신경망(GNN)이 전례 없는 수준의 일반화에 도달하여 재료 발견의 효율성을 10배 향상시킬 수 있음을 보여준다. 기존에 알려진 48,000개의 안정적인 결정을 기반으로 220만 개의 새로운 안정적인 구조를 추가로 발견했으며, 이는 인류에게 알려진 안정적인 재료의 수를 10배 확장한 것이다. 발견된 구조 중 736개는 이미 독립적으로 실험을 통해 구현되었다. 또한, 수억 건의 제일원리 계산을 통해 얻은 방대한 데이터는 이온 전도도의 제로샷 예측과 같은 다운스트림 응용을 위한 고정밀 원자간 전위 모델 개발을 가능하게 했다.
서론
새로운 기능성 무기 재료의 발견은 청정에너지에서 정보 처리 기술에 이르기까지 다양한 기술적 돌파구를 여는 핵심 요소이다. 그러나 실험적 접근은 비용과 시간 문제로 확장이 어렵고, Materials Project와 같은 계산 기반 접근법도 탐색 공간의 방대함으로 인해 한계가 있었다. 특히, 머신러닝 기술은 경쟁 상(phase)에 대한 안정성(분해 에너지)을 정확하게 예측하는 데 어려움을 겪었다. 이 논문은 대규모 액티브 러닝을 통해 재료 탐색을 위한 머신러닝을 확장하여, 안정성을 정확하게 예측하고 재료 발견을 안내할 수 있는 최초의 모델을 개발하는 것을 목표로 한다.
모델 아키텍처 / 방법론
-
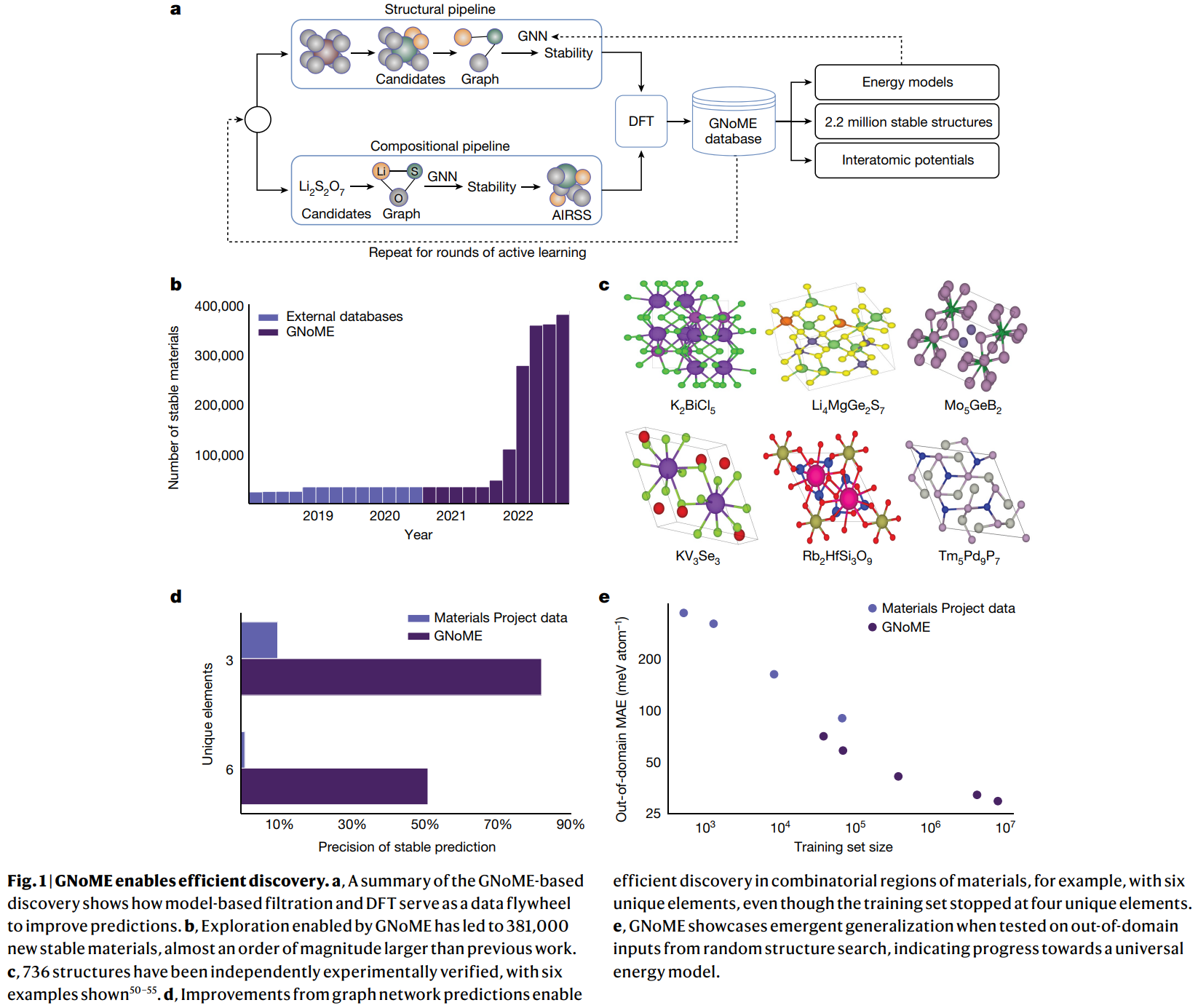
핵심 구조/방법: 연구의 핵심은 GNOME(Graph Networks for Materials Exploration)이라는 액티브 러닝 프레임워크이다. 이 프레임워크는 논문의 Figure 1a에 묘사된 바와 같이, GNN 모델을 이용한 후보군 필터링과 DFT 계산을 통한 검증 및 데이터 추가를 반복하는 '데이터 플라이휠' 구조를 가진다. 이 과정은 두 가지 주요 파이프라인으로 구성된다.
-
주요 구성 요소:
- 구조적 파이프라인(Structural Pipeline): 알려진 결정 구조에서 원자를 치환하여 후보 구조를 생성한다. 특히, 불완전한 치환을 효율적으로 가능하게 하는 새로운 대칭성 인지 부분 치환(Symmetry-Aware Partial Substitutions, SAPS) 기법을 도입하여 후보군의 다양성을 극대화했다.
- 조성적 파이프라인(Compositional Pipeline): 구조 정보 없이 화학 조성만으로 안정성을 예측한다. 유망한 조성에 대해 초기 임의 구조 탐색(Ab Initio Random Structure Searching, AIRSS)을 통해 100개의 초기 구조를 생성하여 평가한다.
- GNN 모델: 구조 또는 조성을 그래프로 변환하여 총 에너지를 예측한다. 이 예측값을 기준으로 DFT 계산을 수행할 유망한 후보군을 선별한다.
- DFT 검증: GNN이 선별한 후보 구조의 에너지를 DFT로 정확하게 계산하여 안정성을 검증하고, 이 결과를 다음 액티브 러닝 라운드의 학습 데이터로 추가하여 모델 성능을 점진적으로 향상시킨다.
-
수식: 머신러닝 원자간 전위(MLIP) 모델 학습에 사용된 손실 함수는 에너지와 힘(force)에 대한 Huber 손실의 가중 합으로 구성된다.
-
알고리즘: 핵심 알고리즘은 다음과 같은 액티브 러닝 순환 과정이다.
- 기존 데이터베이스의 구조를 기반으로 치환(SAPS 포함)을 통해 방대한 수의 후보 구조를 생성한다.
- 학습된 GNN 모델을 사용해 후보들의 안정성을 예측하고 유망한 구조들을 필터링한다.
- 필터링된 구조에 대해 고비용의 DFT 계산을 수행하여 정확한 에너지를 얻는다.
- DFT 계산 결과를 학습 데이터셋에 추가한다.
- 확장된 데이터셋으로 GNN 모델을 재학습시켜 성능을 향상시킨다.
- 이 과정을 6라운드에 걸쳐 반복하여 탐색의 효율성과 정확도를 극대화한다.
실험 결과
- 주요 데이터셋: Materials Project와 OQMD의 2021년 스냅샷 데이터를 초기 데이터로 사용했다. 액티브 러닝을 통해 220만 개의 새로운 안정적인 구조를 포함하는 대규모 데이터셋을 구축했다.
- 핵심 성능 지표:
- 에너지 예측 정확도: 초기 모델의 평균 절대 오차(MAE)는 원자당 21 meV였으나, 6차례의 액티브 러닝을 통해 최종적으로 11 meV까지 감소했다.
- 발견 효율성: 안정적인 재료를 예측하는 정확도(hit rate)가 구조적 파이프라인에서 80% 이상, 조성적 파이프라인에서 33%를 달성하여 기존 연구의 약 1% 대비 월등히 높은 효율을 보였다. 논문의 Figure 1b는 GNOME을 통해 발견된 안정적인 재료의 수가 시간이 지남에 따라 기하급수적으로 증가했음을 보여준다.
- 비교 결과:
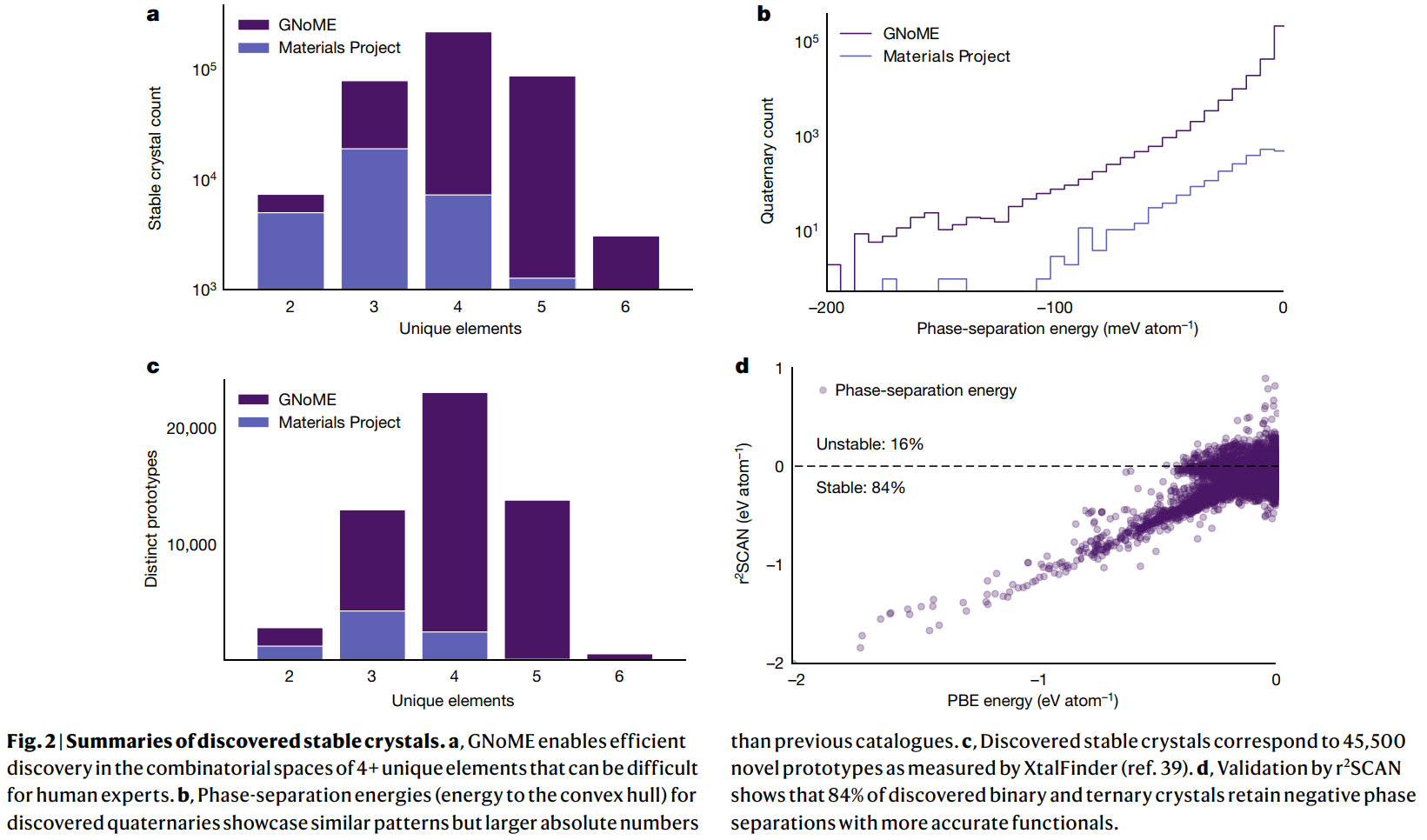
- 신규 발견: 기존 데이터베이스 대비 220만 개의 안정적 구조를 발견했고, 이 중 381,000개는 새로운 볼록 껍질(convex hull) 위의 물질이다. 이는 Figure 2a, c에서 볼 수 있듯이, 특히 4개 이상의 원소로 구성된 복잡한 조성 공간에서 기존보다 훨씬 많은 수의 물질과 새로운 프로토타입을 발견했음을 의미한다.
- 검증: 더 정확한 r²SCAN 함수를 이용한 검증에서, 발견된 2원계 및 3원계 물질의 84%가 안정성을 유지했다. 또한, GNOME이 발견한 구조 중 736개는 ICSD 데이터베이스에 등록된, 독립적으로 실험에서 합성된 물질과 일치함이 확인되었다.
결론
본 연구는 딥러닝 모델의 규모를 확장하는 것이 재료 과학 분야에서 어떻게 전례 없는 발견을 이끌 수 있는지를 명확히 보여준다. GNN과 액티브 러닝을 결합한 GNOME 프레임워크는 220만 개의 새로운 안정적인 결정을 발견하여 알려진 재료의 수를 10배 가까이 늘렸다. 이 연구의 가장 큰 의의는 단순히 많은 수의 재료를 발견한 것을 넘어, 대규모 데이터가 어떻게 머신러닝 모델의 일반화 성능을 향상시켜 학습 분포 밖의 새로운 영역(예: 4개 이상의 원소를 가진 복잡한 화합물)을 탐색하게 하는지를 입증한 데 있다. 또한, 이 과정에서 생성된 데이터셋은 특정 시스템에 대한 추가 학습 없이 범용적으로 사용될 수 있는 강력한 원자간 전위 모델을 탄생시켜, 향후 재료 시뮬레이션 및 설계를 근본적으로 가속화할 것이다.